Overview
The origin of cluster expansions can be traced back to the early 1950s, when Kikuchi developed an Ising model-based cluster variation method to study order-disorder phenomena. In 1984, Sanchez et al. developed a general formalism for the description of configurational cluster expansions in terms of a complete basis set expansion. In simple terms, CE decomposes the energy of a configuration into one-body, two-body, and higher-order interaction terms (‘clusters’), and each term has a corresponding weight called effective cluster interaction (ECI) analogous to the interaction strength. The energy can be exactly reproduced only if all clusters are included in the CE. However, the ECIs for clusters that contain a large number of sites or a large distance between sites are usually negligible. Therefore, the CEs can be truncated to a sum over finite numbers of cluster functions with little loss of accuracy.
The construction of CEs includes data generation by DFT calculations, structure selection and cluster selection. When referring to surface reactions, the training becomes more challenging due to the loss of translational symmetry at the interface and the increase of the complexity of adsorbate interactions and the number of surface species. While DFT calculations typically require a large number of computational resources, it has become more or less standardized in catalysis research. In contrast, the algorithms driving structure selection and cluster selection are arguably more critical to the overall accuracy of the CEs. Therefore, significant efforts have been put into developing approaches to generate effective clusters for a given training set. The goal of the selection algorithm is to find the clusters that have physical contributions to lateral interactions. Early CEs relied on heuristic methods to manually select clusters, while recent approaches incorporate automated ML approaches to systematically optimize the selection process. Multiple automated selection algorithms have been developed for CEs including MIT Ab-initio Phase Stability (MAPS), genetic algorithm (GA), and steepest descent (SD), which incorporate various well-established ML approaches such as cross validation, feature selection, pattern recognition, and regularization.
Here I employed an artificial neural network called CE-TinNet to predict the cluster expansions of the studied structures and predict the formation/adsorption energies. As a benchmark, I also used a previous model LCNN developed by Lym et. al, and the state-of-the-art CGCNN model to make a prediction for the same property.
Lattice convolution neural network
The lattice convolutional neural network (LCNN) is a Python library for deep learning of lattice system developed by the Vlachos group at the University of Delaware and Jung group at KAIST. The model has been built in hope to improve upon cluster expansion methods, linear regression based on the clusters in the lattice. The LCNN performs better than the cluster expansion and cluster expansion based machine learning methods with sufficient number of data points.
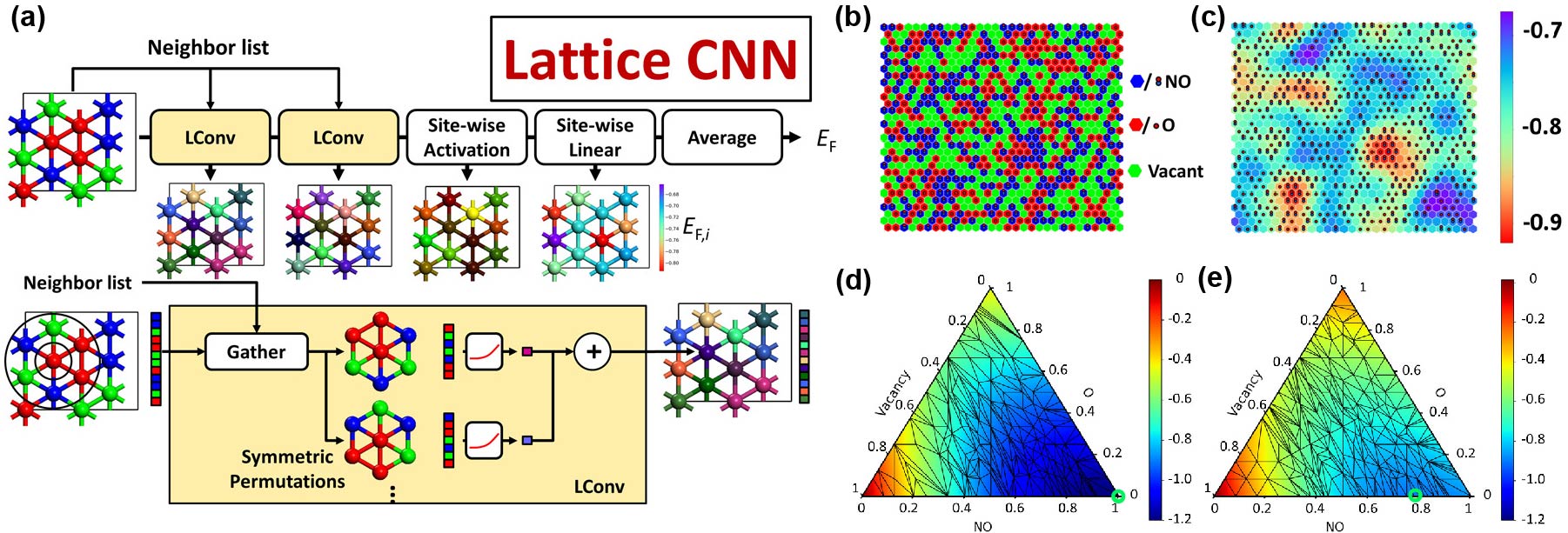
Lym et al. developed a novel lattice convolutional neural network (LCNN) that improved the formation energy prediction compared to state-of-the-art CE methods by 20-30% (Figure a). Featurization process extracts features of each site’s neighbor lists using one-hot encoding. The extracted information then undergoes lattice convolutional (LConv) layers which are built to construct the local environment by considering the nearest neighbors and including permutations to account for the symmetry of the lattice. Activation and summation are followed for new site features. Once all convolution operations are performed, the representation of the lattice is obtained. Followed by the site-wise activation and linear multiplication, the formation energy of each image can be predicted. They applied the LCNN model to study the coverage of O and NO adsorption on Pt(111) surface, the model captures the local environment of adsorbates, and a one-hot encoding input and the predicted site formation energy distribution are shown in Figure b and c, respectively. Compared to CE models, the LCNN achieves the best performance with a test root mean squared error (RMSE) of 4.4 meV/site using less training data. The effect of van der Waals (vdW) forces on lateral interactions have also been studied by comparing the formation energy with and without vdW forces for an example of all unique configurations in a unit cell containing up to 12 adsorption sites (1,581,607 in total). The convex hull diagrams of LCNN computed formation energies trained using PBE-D3 (with vdW forces) and PBE (without vdW forces) datasets are shown in Figure d and e, respectively. They found that the trend in the convex hull has changed by the vdW interactions. Particularly, the global ground state configuration for PBE is observed at 0.78 monolayer (ML) of NO, whereas it is 1.0 ML of NO for PBE-D3.
Crystal Graph Convolutional Neural Networks
The use of machine learning methods for accelerating the design of crystalline materials usually requires manually constructed feature vectors or complex transformation of atom coordinates to input the crystal structure, which either constrains the model to certain crystal types or makes it difficult to provide chemical insights. Thus, they developed a crystal graph convolutional neural networks framework to directly learn material properties from the connection of atoms in the crystal, providing a universal and interpretable representation of crystalline materials. Their method provides a highly accurate prediction of density functional theory calculated properties for eight different properties of crystals with various structure types and compositions.
Dataset
All the data used to train, validate, andtest the machine-learning models on the Pt(111) system wasprovided by Bajpai et al. The formation energy per site is calculated by:
\(
\begin{align}
\begin{split}
Pt + \frac{\theta_O}{2}O_2 + \theta_{NO}NO \rightarrow PtO_{\theta_O}NO_{\theta_{NO}} \> \> \> (\sigma)
\end{split}
\end{align}
\)
\(
\begin{align}
\begin{split}
E_F(\sigma) = \frac{E_0(\sigma)-E_0(0)}{N} - \frac{\theta_O}{2}E_{0,O_2} - \theta_{NO}E_{0,NO}
\end{split}
\end{align}
\)
The Pt(111) surface can be covered by NO, O adsorbate with 600 data points in total, and the unit cell is expanded when there are more adsorbates or at high coverages.
The details of data preprocessing can be found in my previous post
Results and discussion
CE-TinNet is a ML based model that considers the lateral interactions between the adsorbates and surface atoms. It outperforms the LCNN when considering the adsorbate-adsorbate interactions for NO and O adsorption on Platinum.
More studies have been down on different surfaces with various adsorbates, and I’ll share more results when it’s ready to be published.
Tianyou Mou
Chemical Engineer and Data Scientist

Comments