Background
The evolution of the research workflow in computational chemistry is about 3 generations. The first-generation approach is to calculate the physical properties of an input structure, which is often performed via an approximation to the Schrödinger equation
For example, Density functional theory. In the second-generation approach, by using global optimization (for example, an evolutionary algorithm), an input of chemical composition is mapped to an output that contains predictions of the structure or ensemble of structures
that the combination of elements are likely to adopt. The emerging third-generation approach is to use machine-learning techniques with the ability to predict composition, structure and properties. However, you have to provide sufficient data and an appropriate model.
Also, there are four stages of training a machine-learning model: Data collection, Representation(like preprocessing), type of learning, and Model selection. A very important factor is that you have to provide sufficient amount of accurate data to train your machine learning model.
With years of hard work, many DFT computational databases have been setup. Most of them are very large. But the problem is most calculations are calculated by basic exchange correlation functionals, like gga. So the accuracy is relative low. At the same time, there are some experimental databases with very high accuracy. But since lots of cases you couldn’t really conduct the experiments. So the size of the dataset is limited. We can also obtain some other quantum-mechanical databases like coupled cluster quantum method database. Although they have a better accuracy, all of them are computational expensive.
So, right now we don’t have a proper database with high accuracy and large size.
We can take a look at some examples. OQMD is A large dataset containing DFT-computed materials properties for 341K materials. But Thermo-Calc Software is An experimental data set containing only 1963 samples. So, here comes the question: machine learning needs large amount of data to be accurate, can we use limited amounts of data to achieve a high accuracy?
There is a Machined Learning framework called “ transfer learning ” which has considerable potential to overcome the problem of limited amounts of data. Transfer learning relies on the concept that various property types, such as physical, chemical, electronic, thermodynamics are physically interrelated.
Previously, if we want to predict a target property with small dataset, we just train this small dataset, although the dataset is accurate, with limited amount of data, we couldn’t get a good accuracy. For transfer learning, we can use our exist big dataset to train a model first, and save the common features that are same for different materials, and then use this model to train another small dataset, and then predict a target property with a higher accuracy.
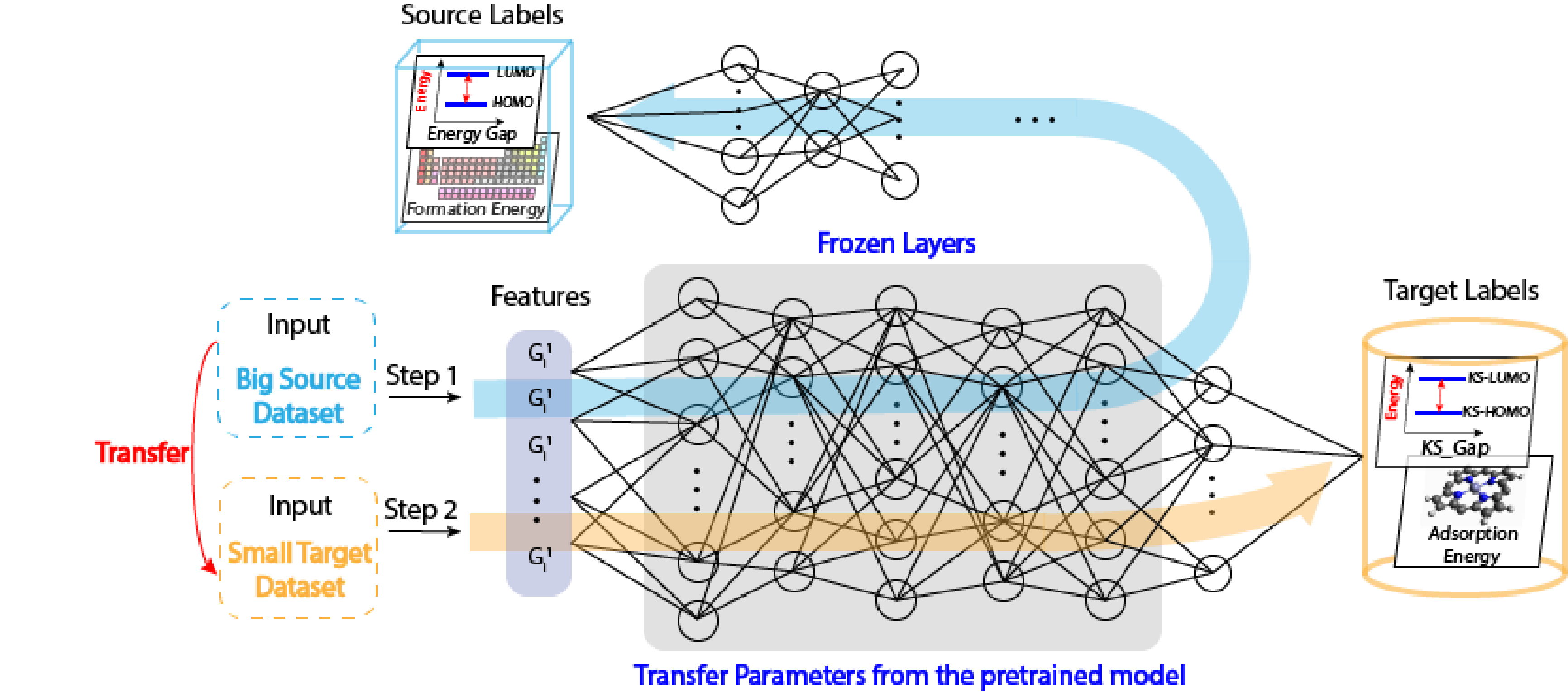
Then, let’s focus on how does this transfer learning work. There are two commonly applied procedures, one is frozen featurizer, another one is called fine-tuning techniques. I’m gonna mainly talk about this fine-tuning techniques. We still have two steps, the first step is using big dataset to pretrain a model. The second step is using this model to predict a target property. We first input big data, and here we train this deep neural networks, common features are saved in these layers. Then we can use this model, but freeze those layers since we don’t want to lose those common features. We just let the last several layers to be optimized. Then we can get a good prediction of our target property.
Models
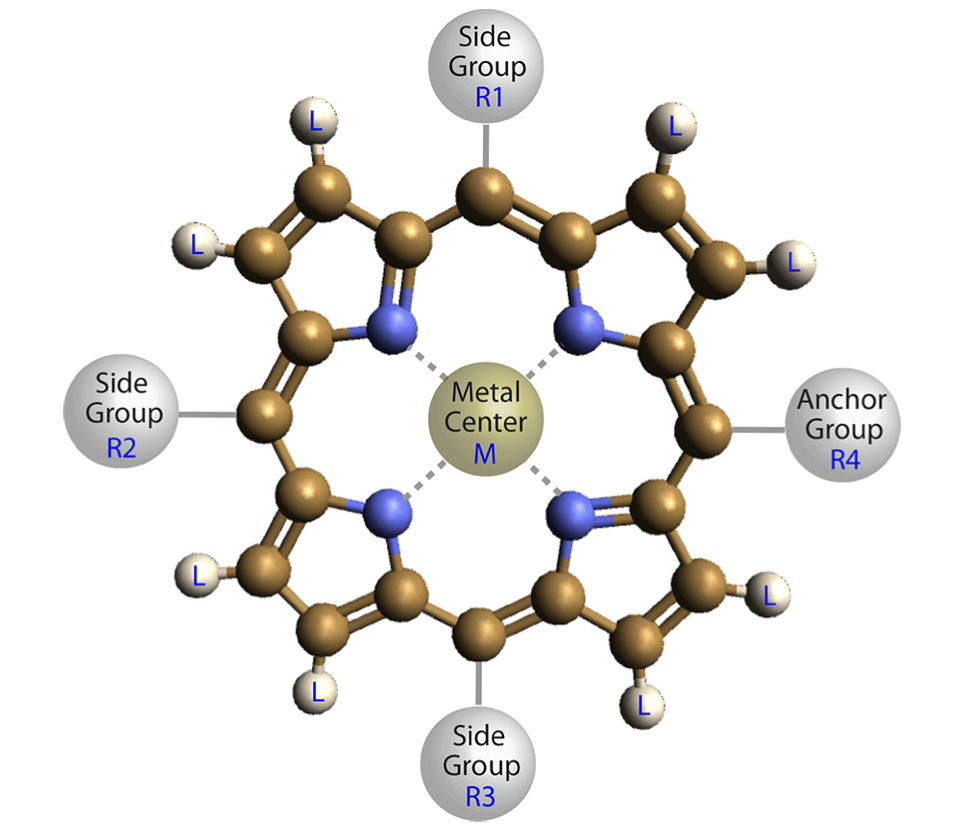
The data set for machine-learning was retrieved from the Computational Materials Repository, which includes > 12000 molecular structures of porphyrins and DFT-calculated properties. The structure of the porphyrin is shown below.
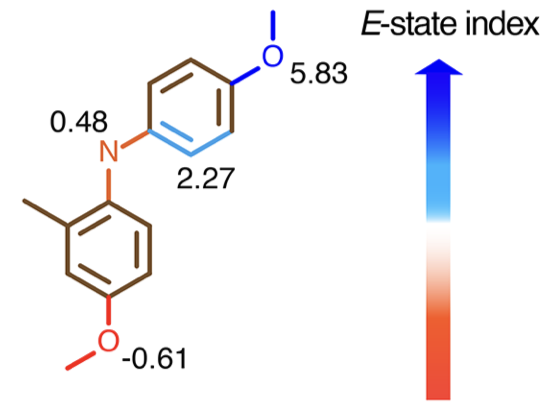
Molecular representation, or feature engineering is a key ingredient in the development of machine-learning models. Here, I employed the electrotopological-state index (E-state index), which has been widely used in quantitative structure-property relationship (QSPR) models to predict boiling points, aqueous solubilities, and other thermophysical properties of organic molecules.
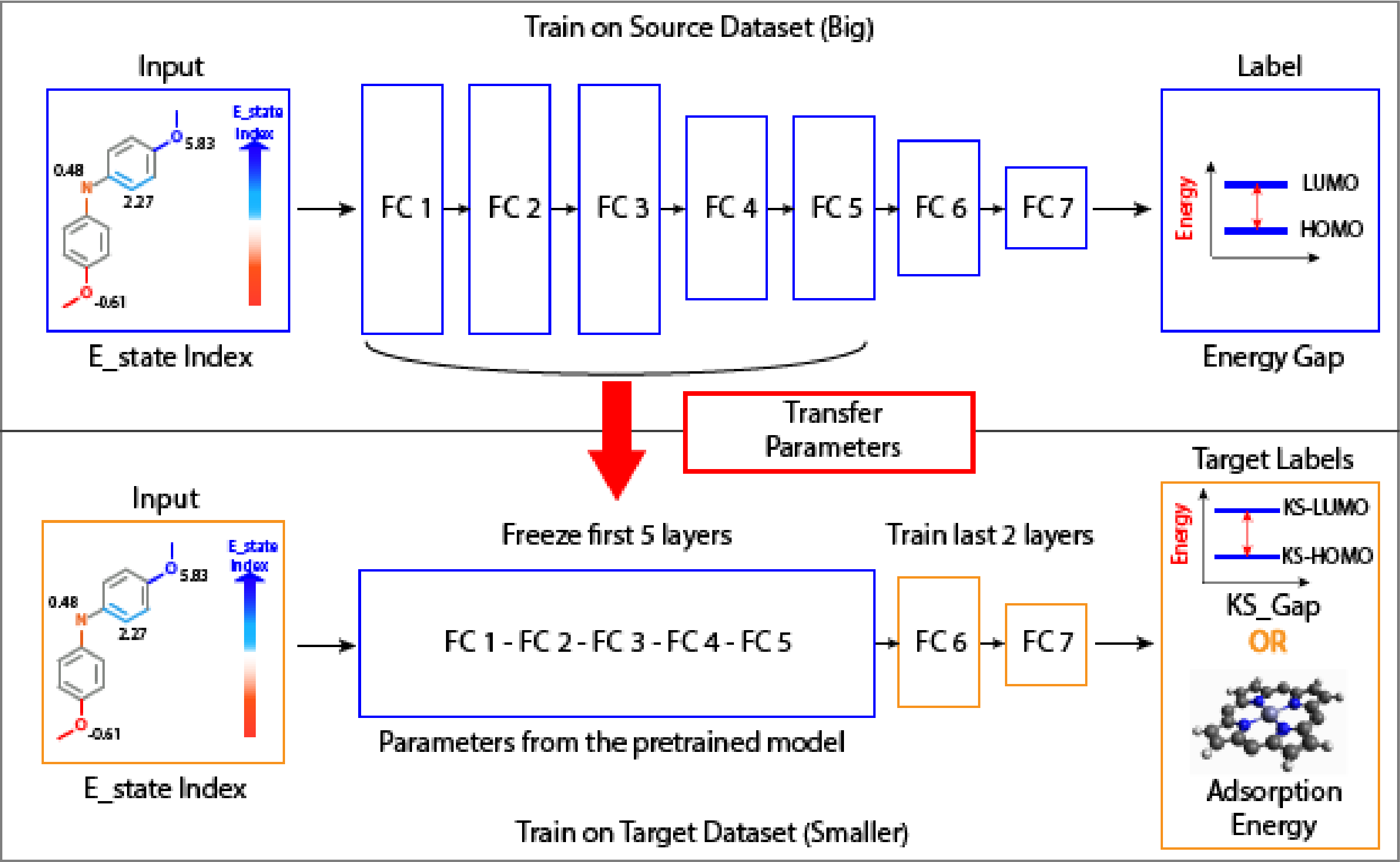
A deep neural network model called ElemNet is used to conduct the transfer learning framework. It has multiple layers of fully connected layers, some of which can be frozen for the training of transfer learning.
The schematic flow of the ElemNet-TL model is shown below, which uses the E-state index as the input to train a big source data to predict a target property. The pertained model is then employed with first couple layers fixed, and the target small dataset is used to train the last several layers and then predict another target property.
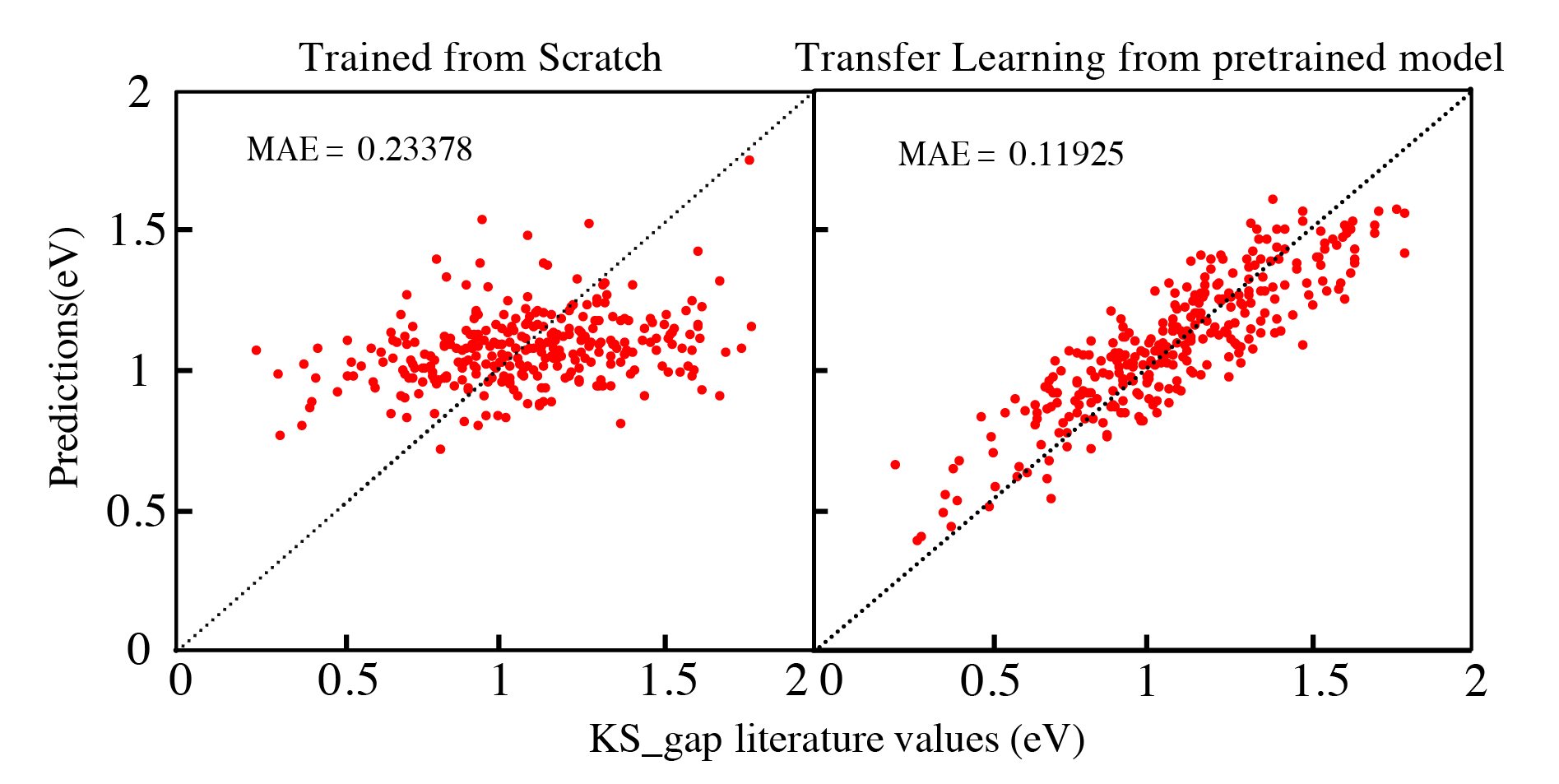
At last, we can compare the result of training from scratch and training with transfer learning, we can see with transfer learning applied, we can get a much better performance even though we both just use the same experimental dataset to train the model. That’s because the transfer learning method use the pretrained large dataset model first, and save the common features in the hidden layers. This helps to improve the performance.
Conclusion
In summary, A transfer learning model shows an enhanced performance compared with traditional machine learning model
Also, Transfer learning can be further applied to other area with limited amount of accurate data.
References
1.Jha, D.; Ward, L.; Paul, A.; Liao, W.-K.; Choudhary, A.; Wolverton, C.; Agrawal, A. ElemNet: Deep Learning the Chemistry of Materials From Only Elemental Composition. Sci. Rep. 2018, 8 (1), 17593.
2.Li, Z.; Omidvar, N.; Chin, W. S.; Robb, E.; Morris, A.; Achenie, L.; Xin, H. Machine-Learning Energy Gaps of Porphyrins with Molecular Graph Representations. J. Phys. Chem. A 2018, 122 (18), 4571–4578.
Tianyou Mou
Chemical Engineer and Data Scientist

Comments